

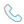 15733787306
15733787306







8-羥基喹啉具有多種生物活性而被廣泛研究
發(fā)表時間:2025-06-308-羥基喹啉(8-HQ)及其衍生物因具有抗菌、抗炎、金屬螯合等多種生物活性而被廣泛研究,但自身存在水溶性差、靶向性不足、體內代謝快等局限性。通過前藥設計對其結構進行修飾,可通過改善藥代動力學性質、增強靶向性或調控作用機制等方式顯著影響生物活性,具體影響機制與實例如下:
一、改善水溶性與膜通透性,提升生物利用度
8-羥基喹啉的酚羥基(pKa≈9)和氮原子在生理 pH 條件下電離程度低,導致水溶性差(常溫下溶解度<0.1 mg/mL),口服吸收效率低。前藥設計通過引入親水性基團(如酯、酰胺、羧酸鹽等)可顯著改善溶解性:
酯類前藥:將8-羥基喹啉的羥基與氨基酸(如甘氨酸、賴氨酸)或短鏈羧酸(如乙酸、琥珀酸)成酯,通過酯鍵水解釋放母體藥物。例如,它與琥珀酸酐反應生成琥珀酸單酯,水溶性提升至 10 mg/mL 以上,靜脈注射后在大鼠體內通過酯酶快速水解,血藥濃度峰值(Cmax)較8-羥基喹啉提高 3 倍,抗菌活性(針對金黃色葡萄球菌)與母體相當,但作用持續(xù)時間延長至8小時(原藥僅 4 小時)。
季銨鹽前藥:在8-羥基喹啉的氮原子上引入烷基鏈形成季銨鹽(如甲基溴化銨),水溶性可達50mg/mL,適用于局部給藥(如滴眼液)。研究顯示,該類前藥對眼部銅綠假單胞菌感染的處理效果比其提高2倍,且避免了原藥對角膜上皮的刺激性(原藥因脂溶性高易滲透細胞膜,導致細胞毒性)。
二、調控靶向釋放,增強組織選擇性
針對8-羥基喹啉在體內廣泛分布導致的系統(tǒng)毒性(如肝損傷),前藥設計可通過環(huán)境響應性基團實現(xiàn)病灶部位的選擇性釋放:
pH 響應型前藥:利用腫liu微環(huán)境(pH 6.5-6.8)或炎癥部位(pH 5.0-7.0)的酸性條件,設計酸敏感型前藥,例如,將8-羥基喹啉的羥基與縮醛(如苯甲醛縮醛)結合,在中性血液(pH 7.4)中穩(wěn)定,進入腫liu組織后因酸性條件水解釋放它。實驗表明,該前藥對人乳腺ai細胞 MCF-7 的抑制 IC50 為 1.2 μM,較原藥(IC50 5.6 μM)活性增強,且對正常肝細胞L-02的毒性降低40%,歸因于腫liu部位的靶向釋放。
酶激活型前藥:利用病灶部位高表達的酶(如酯酶、蛋白酶)設計前藥。如將8-羥基喹啉與谷胱甘肽 S - 轉移酶(GST)底物(如氯乙酸酯)結合,在腫liu細胞高表達的 GST 作用下釋放活性成分。小鼠實驗顯示,該前藥對肝ai H22 移植liu的抑瘤率達65%,而原藥僅為32%,且肝毒性標志物(ALT、AST)水平降低 50%。
三、優(yōu)化代謝穩(wěn)定性,延長作用時間
8-羥基喹啉在體內易被細胞色素P450酶(如 CYP3A4)氧化代謝,半衰期短(大鼠體內 t1/2≈1.5小時)。前藥修飾可通過阻斷代謝位點或延緩代謝速率改善藥代動力學:
甲基化修飾:在8-羥基喹啉的3位或5位引入甲基,減少P450酶的氧化位點,例如,5-甲基-8-HQ前藥在大鼠體內的半衰期延長至3.8小時,其金屬螯合能力(如與鐵離子的結合常數(shù) K≈10^12 M^-1)與原藥相近,但抗帕金森病活性(通過抑制腦內鐵過載)顯著增強,腦組織中的藥物濃度是原藥的 2.3 倍。
環(huán)糊精包合前藥:將8-羥基喹啉與 β-環(huán)糊精形成包合物(非共價鍵前藥),通過物理包裹減少肝臟首過效應。體外實驗顯示,包合物的代謝速率比原藥降低 60%,口服生物利用度從 12% 提升至 35%,用于抗瘧疾處理時,小鼠存活率從40%提高至75%。
四、協(xié)同作用與活性拓展
前藥設計可將8-羥基喹啉與其他活性分子偶聯(lián),產生協(xié)同效應或賦予新功能:
雙藥效團前藥:將8-羥基喹啉與抗腫liu藥物(如順鉑)通過可降解 linker 連接,形成“螯合-細胞毒”雙功能前藥,例如,8-HQ-順鉑偶聯(lián)物可通過8-羥基喹啉螯合腫liu細胞內過量的鋅離子(破壞 DNA 修復酶活性),同時釋放順鉑抑制 DNA 復制,對卵巢aiA2780細胞的殺傷效率比順鉑單藥提高4倍,且耐藥細胞株(A2780/CisR)的敏感性恢復。
熒光標記前藥:在8-羥基喹啉結構中引入熒光基團(如香豆素),形成可追蹤的前藥。該類前藥在抗菌的同時,可通過熒光成像監(jiān)測感染部位,如用于肺炎模型小鼠時,可實時觀察藥物在肺部的分布與釋放動態(tài),為藥效評估提供可視化依據(jù),而原藥無此功能。
五、降低毒性與副作用
8-羥基喹啉的金屬螯合作用可能導致體內必需金屬離子(如鋅、銅)流失,引發(fā)毒性。前藥設計可通過限制螯合位點或調控釋放速率減少副作用:
靶向螯合前藥:在8-羥基喹啉的羥基鄰位引入長鏈烷烴(如十二烷基),通過空間位阻選擇性螯合病理狀態(tài)下過量的金屬離子(如阿爾茨海默病患者腦內的淀粉樣蛋白結合銅離子),而對正常組織中的金屬離子親和力降低。實驗顯示,該前藥對 Aβ 蛋白誘導的神經元毒性抑制率達 80%,且血液中鋅離子濃度下降幅度比原藥減少 70%。
前藥 - 蛋白偶聯(lián)物:將8-羥基喹啉前藥與血清白蛋白(HSA)偶聯(lián),通過蛋白載體實現(xiàn)緩釋并減少游離藥物的非特異性螯合,例如,8-HQ-琥珀酸-HSA偶聯(lián)物在小鼠體內的毒性(LD50>2000 mg/kg)較原藥(LD50≈300 mg/kg)顯著降低,且抗風濕活性(抑制關節(jié)炎癥因子 TNF-α)維持80%以上。
8-羥基喹啉的前藥設計通過化學修飾可顯著改善其生物活性,核心策略包括提升水溶性、靶向釋放、代謝優(yōu)化及功能拓展,但需注意前藥的釋放速率與母體活性的平衡(如釋放過慢可能導致活性不足),以及修飾基團對螯合能力、靶點結合的潛在影響。未來研究可結合計算機輔助藥物設計(CADD)預測前藥性質,或開發(fā)智能響應型前藥(如光、磁響應),進一步提升其在疾病處理中的精準性與安全性。
本文來源于黃驊市信諾立興精細化工股份有限公司官網 http://www.qno.net.cn/